spammR Spatial Proteomics Example
Sara Gosline
Jul 08, 2026
Source:vignettes/spatProt.Rmd
spatProt.RmdGetting started
To install the package currently you must install directly from
GitHub along with the leapR dependency as shown below.
Before release we hope to move to Bioconductor.
The leapR package is designed for flexible pathway
enrichment and currently must be installed before spammR.
##install if not already installed
library(devtools)
devtools::install_github('PNNL-CompBio/leapR')
devtools::install_github('PNNL-CompBio/spammR')Once the package is installed you load the library, including the test data.
Collecting data to analyze
spammR enables the analysis of disparate sets of
multiomic data: image-based data and numerical measurements of omics
data. It is incredibly flexible as to the type of multiomic
data. We assume each omics measurement is collected in a single sample,
and that there are specific spatial coordinates for that sample in the
image. We leverage the SpatialExperiment object to store
the data for each image/measurement pair.
Data overview and examples
The spammR package requires omics data with spatial
coordinates for the functions to run successfully. Here we describe the
data required and show examples.
Omics Measurement Data
SpatialExperiment can hold multiple omics measurements
mapping to the same sample identifier in different ‘slots’. This data
can be a tabular data frame or matrix with rownames referencing
measurements in a particular sample (e.g. gene, species) and column
names representing sample identifiers. An example of this can be found
by loading pancDataList.rda file from Figshare.
To evaluate the features of this package we are using pancreatic data from Gosline et al. that is captured using mass spectrometry measured from 7 independent regions of a single human pancreas. Each image is segmented into nine ‘voxels’, with one voxel per image representing a cluster of islet cells.
path <- tempfile()
bfc <- BiocFileCache(path, ask = FALSE)
pdl_f <- "https://api.figshare.com/v2/file/download/55158821"#,
# mode = "wb", quiet = TRUE, dest = "pdl.rda")
pc <- bfcadd(bfc, "pdl", fpath=pdl_f)## Error while performing HEAD request.
## Proceeding without cache information.## 0_S_1_1 0_S_1_2 0_S_1_3 0_S_2_1 0_S_2_2 0_S_2_3
## sp|A0A024RBG1|NUD4B_HUMAN 13.06042 13.42317 12.42396 13.02470 12.56442 12.69023
## sp|A0A096LP55|QCR6L_HUMAN 15.10920 15.27460 15.16780 15.01030 15.46639 14.73712
## sp|A0AV96|RBM47_HUMAN 17.40246 17.29727 17.25559 17.34851 17.12866 17.17658
## sp|A0AVT1|UBA6_HUMAN 18.00653 18.43015 18.24663 18.17563 18.38961 18.29268
## sp|A0FGR8|ESYT2_HUMAN 16.59018 16.48890 16.50134 16.55334 16.32316 16.43397
## sp|A0MZ66|SHOT1_HUMAN 18.19277 18.73633 18.54485 18.20005 18.74041 18.70762
## 0_S_3_1 0_S_3_2
## sp|A0A024RBG1|NUD4B_HUMAN NA 13.40670
## sp|A0A096LP55|QCR6L_HUMAN 14.81792 15.63741
## sp|A0AV96|RBM47_HUMAN 17.27792 17.11678
## sp|A0AVT1|UBA6_HUMAN 18.12080 18.10220
## sp|A0FGR8|ESYT2_HUMAN 16.43682 16.20515
## sp|A0MZ66|SHOT1_HUMAN 18.60198 18.62946
file.remove("pdl.rda")## Warning in file.remove("pdl.rda"): cannot remove file 'pdl.rda', reason 'No
## such file or directory'## [1] FALSEHere the rownames represent protein identifiers and the column names represent individual samples. Each element of the list contains the measurements from a different sample:
## [1] 7
head(pancDataList[[2]][, 1:8])## 1_S_1_1 1_S_1_2 1_S_1_3 1_S_2_1 1_S_2_2 1_S_2_3
## sp|A0A024RBG1|NUD4B_HUMAN NA NA NA NA NA NA
## sp|A0A096LP55|QCR6L_HUMAN NA NA NA NA NA NA
## sp|A0AV96|RBM47_HUMAN 17.68866 17.58076 17.51335 17.65900 17.52926 17.44999
## sp|A0AVT1|UBA6_HUMAN 18.02235 18.35493 18.03651 18.09462 18.08796 18.04204
## sp|A0FGR8|ESYT2_HUMAN 17.50177 17.51525 17.34338 17.41622 17.34254 17.46062
## sp|A0MZ66|SHOT1_HUMAN 18.57445 18.67262 18.83672 18.69094 18.48785 18.75190
## 1_S_3_1 1_S_3_2
## sp|A0A024RBG1|NUD4B_HUMAN NA NA
## sp|A0A096LP55|QCR6L_HUMAN NA NA
## sp|A0AV96|RBM47_HUMAN 17.37103 17.50367
## sp|A0AVT1|UBA6_HUMAN 18.21052 17.94707
## sp|A0FGR8|ESYT2_HUMAN 17.31495 17.53217
## sp|A0MZ66|SHOT1_HUMAN 18.82805 18.48856This list is used below in our analysis examples.
Sample metadata
The samples metadata table contains mappings between samples and
metadata. An example can be found in data(pancMeta). Most
importantly we require the image mapping information, which includes: -
Image coordinates: to map the image to a coordinate space we
need to know the x_origin, and y_origin
(assumed to be zero) as well as x_max and
y_max, which is the top right of the image. The package
plots the entire image so specifying these coordinates ensures
that all other points are properly mapped. - Sample
coordinates: Each sample has its own x_coord and
y_coord. - Spot size: spot_height and
spot_width.
## Image x_coord y_coord IsletStatus IsletOrNot Plex Grid.Number x_pixels
## 0_S_3_1 0 3 1 Proximal NonIslet 127N 1 475
## 0_S_2_1 0 2 1 Islet Islet 128N 2 380
## 0_S_1_1 0 1 1 Proximal NonIslet 127C 3 285
## 0_S_3_2 0 3 2 Proximal NonIslet 128C 4 475
## 0_S_2_2 0 2 2 Proximal NonIslet 129N 5 380
## 0_S_1_2 0 1 2 Proximal NonIslet 129C 6 285
## y_pixels x_origin y_origin x_max y_max spot_width spot_height
## 0_S_3_1 170 0 0 860 725 90 140
## 0_S_2_1 170 0 0 860 725 90 140
## 0_S_1_1 170 0 0 860 725 90 140
## 0_S_3_2 315 0 0 860 725 90 140
## 0_S_2_2 315 0 0 860 725 90 140
## 0_S_1_2 315 0 0 860 725 90 140This metadata contains information for all 7 images, so we do not
need a separate metadata file for each image, the
convert_to_spe function will simply take the metadata
relevant to the data file.
Image files
There can be multiple image files associated with a single set of
omics measurements. Currently we have tested working with files in
png format. Each image we have is stained so that we can
identify the Islet cells. Each image also has a grid superimposed to
show where the sample measurements came from. The grid is not necessary,
of course, but can help alibrate the coordinates.
library(cowplot)
cowplot::ggdraw() + cowplot::draw_image(system.file("extdata",
"Image_1.png",
package = "spammR"))Now we can use this image and others to visualize omics data.
Omics metadata
The last set of metadata relates to the rows of the
omics measurement data. When using gene-based data, this will be the
genes or proteins in the dataset. When using metagenomics, this will
refer to the species. One column of this table must uniquely map to the
rownames of the omics data.
## pancProts EntryName PrimaryGeneName
## 1 sp|A0A024RBG1|NUD4B_HUMAN NUD4B_HUMAN NUDT4B
## 2 sp|A0A096LP55|QCR6L_HUMAN QCR6L_HUMAN UQCRHL
## 3 sp|A0AV96|RBM47_HUMAN RBM47_HUMAN RBM47
## 4 sp|A0AVT1|UBA6_HUMAN UBA6_HUMAN UBA6
## 5 sp|A0FGR8|ESYT2_HUMAN ESYT2_HUMAN ESYT2
## 6 sp|A0MZ66|SHOT1_HUMAN SHOT1_HUMAN SHTN1This data helps us find better gene identifiers.
Loading data into spatial experiment object.
Now that we have all the data loaded we can build a
SpatialExperiment object either using ALL samples or just
the samples in a single image. We can pool all the data for more
statistical power.
pooledData <- dplyr::bind_cols(pancDataList)
pooled.panc.spe <- convert_to_spe(pooledData, ## pooled data table
pancMeta, ## pooled metadata
protMeta, ## protein identifiers
feature_meta_colname = "pancProts", # column name
)## Spatial object created without spatial coordinate
## column names provided. Distance based analysis will not be enabled.## Note: Only mapping metadata for 6662 features out of 6693 data points
print(pooled.panc.spe)## class: SpatialExperiment
## dim: 6662 63
## metadata(0):
## assays(1): proteomics
## rownames(6662): sp|A0A024RBG1|NUD4B_HUMAN sp|A0A096LP55|QCR6L_HUMAN ...
## sp|Q9Y3M8|STA13_HUMAN sp|Q9Y6X3|SCC4_HUMAN
## rowData names(6): pancProts Entry ... GeneNames PrimaryGeneName
## colnames(63): 0_S_1_1 0_S_1_2 ... 3_S_3_2 3_S_3_3
## colData names(16): Image x_coord ... spot_height sample_id
## reducedDimNames(0):
## mainExpName: NULL
## altExpNames(0):
## spatialCoords names(0) :
## imgData names(0):We can also create a list of SpatialExperiment objects,
one for each of the 3 images we have.
Now we can use these individual image objects or the combined ‘pooled’ object for analysis.
Spatial data with image
Here we loop over all of the images in imglist to plot
the expression of the insulin protein in each image. We expect insulin
(or INS) to be highest in voxels containing islet cells, which we label
using the label_column ‘IsletOrNot’ which was loaded into
the metadata for us.
allimgs <- lapply(imglist, function(x) {
spe <- img.spes[[x]]
res <- spatial_heatmap(spe,
feature = "INS",
feature_type = "PrimaryGeneName",
sample_id = x,
image_id = "with_grid",
label_column = "IsletOrNot",
interactive = FALSE
)
return(res)
})
allimgs[[2]]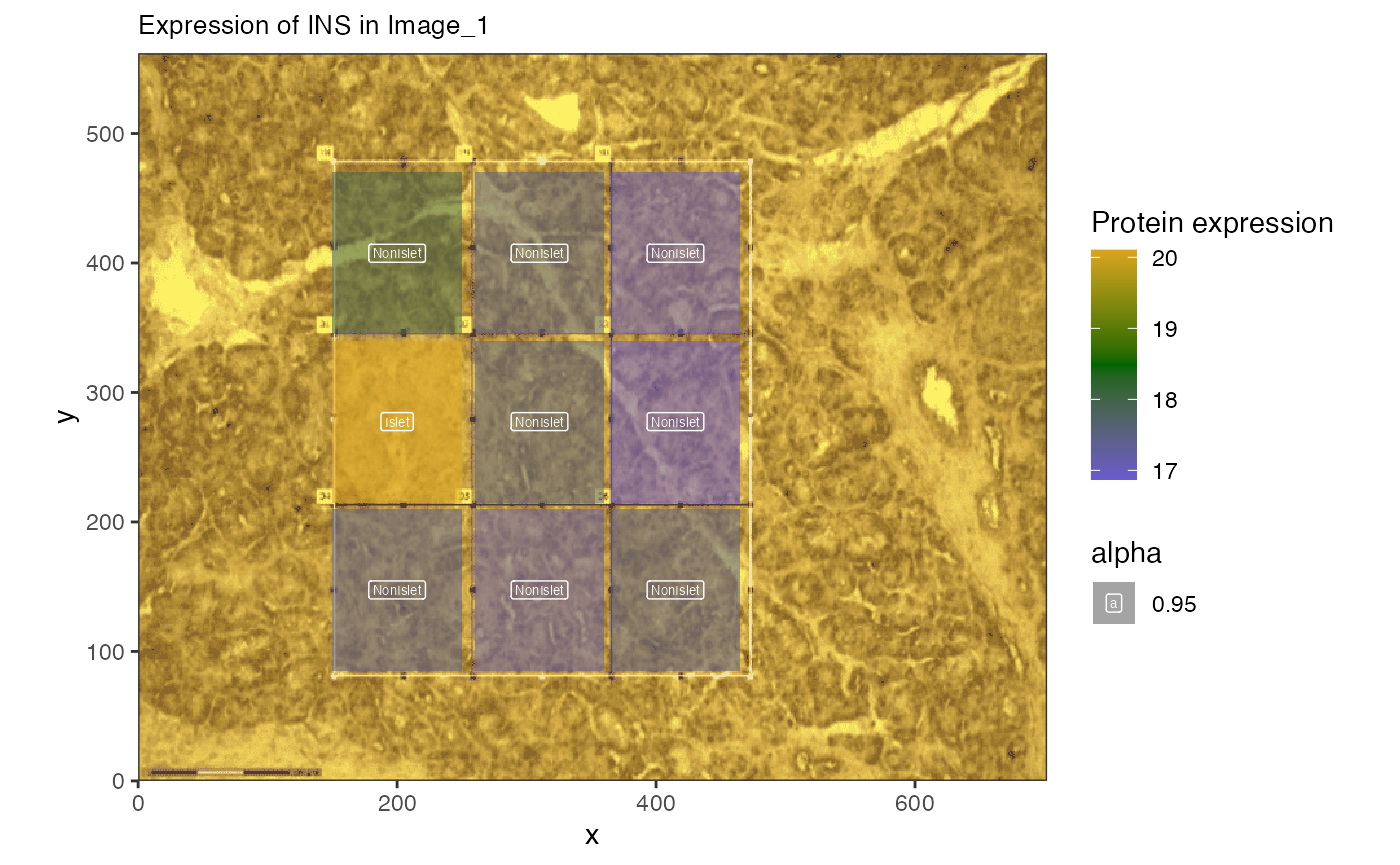
To go further and visualize entire pathways we need to first identify which groups of proteins are of interest using a more unsupervised approach.
Expression and pathway analysis
Now that we have the ability to overlay omic measurements with image
ones, we can identify new features to plot and visualize them. First we
can employ standard differential expression approaches using the voxel
labels and the limma pathway.
Differential expression
First we want to identify specific proteins that are up-regulated in the islet cells (or regions labeled ‘islet’) compared to other regions. We can then plot the set of proteins.
islet_res <- calc_spatial_diff_ex(pooled.panc.spe,
assay_name = "proteomics",
log_transformed = FALSE,
category_col = "IsletOrNot"
)
# we filter the significant proteins first
sig_prots <- subset(rowData(islet_res),
NonIslet_vs_Islet.adj.P.Val.limma < 0.01)
# then separate into up-regulated and down-regulated based on fold chnage
ups <- subset(sig_prots, NonIslet_vs_Islet.logFC.limma > 0)
downs <- subset(sig_prots, NonIslet_vs_Islet.logFC.limma < 0)
print(paste(
"We found", nrow(sig_prots), "significantly differentally \
expressed proteins including",
nrow(ups), "upregulated proteins and", nrow(downs), "downregulated"
))## [1] "We found 241 significantly differentally \n expressed proteins including 168 upregulated proteins and 73 downregulated"Now we can plot those differentially expressed proteins across images.
Plot differentially expressed proteins
If we are interested in the combined expression of proteins we can also visualize those.
spe.plot <- img.spes[[2]]
hup <- spatial_heatmap(spe.plot,
feature = rownames(downs),
sample_id = "Image_1",
image_id = "with_grid",
label_column = "IsletOrNot",
interactive = FALSE
)
hup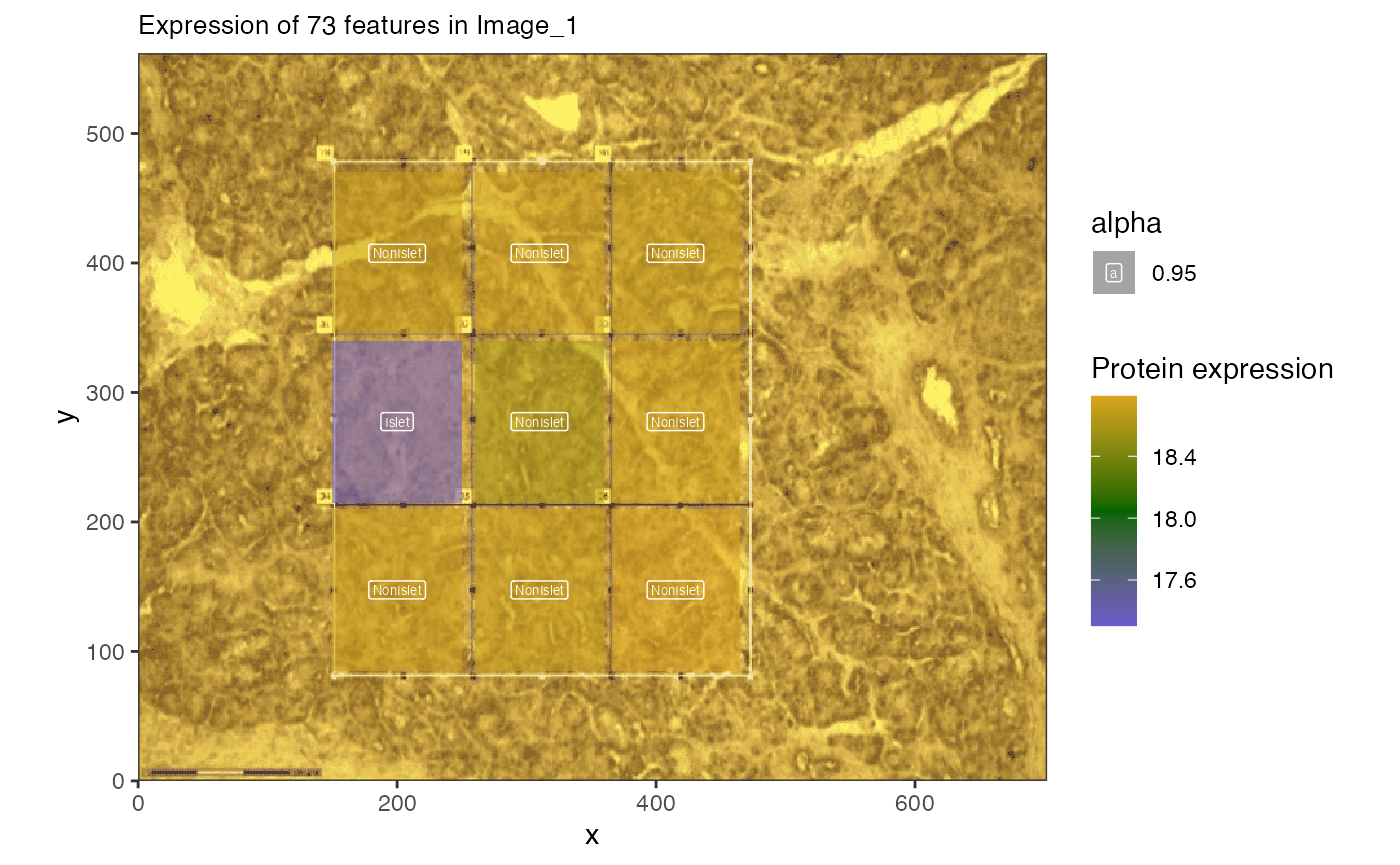
Pathway enrichment measurements
Now we can calculate the enriched pathways in the islets.
library(leapR)
data("krbpaths")
ora.res <- enrich_ora(islet_res, geneset = krbpaths,
geneset_name = "krbpaths",
feature_column = "PrimaryGeneName")
print(ora.res[grep("INSULIN", rownames(ora.res)),
c("ingroup_n", "pvalue", "BH_pvalue")])## ingroup_n
## KEGG_INSULIN_SIGNALING_PATHWAY 5
## BIOCARTA_INSULIN_PATHWAY 2
## REACTOME_GLUCOSE_REGULATION_OF_INSULIN_SECRETION 13
## REACTOME_INSULIN_SYNTHESIS_AND_SECRETION 24
## REACTOME_REGULATION_OF_INSULIN_LIKE_GROWTH_FACTOR_ACTIVITY_BY_INSULIN_LIKE_GROWTH_FACTOR_BINDING_PROTEINS 0
## REACTOME_REGULATION_OF_INSULIN_SECRETION 16
## REACTOME_REGULATION_OF_INSULIN_SECRETION_BY_ACETYLCHOLINE 7
## REACTOME_REGULATION_OF_INSULIN_SECRETION_BY_GLUCAGON_LIKE_PEPTIDE_1 11
## REACTOME_REGULATION_OF_INSULIN_SECRETION_BY_FREE_FATTY_ACIDS 7
## REACTOME_INHIBITION_OF_INSULIN_SECRETION_BY_ADRENALINE_NORADRENALINE 4
## pvalue
## KEGG_INSULIN_SIGNALING_PATHWAY 1.000000e+00
## BIOCARTA_INSULIN_PATHWAY 2.247236e-01
## REACTOME_GLUCOSE_REGULATION_OF_INSULIN_SECRETION 5.549704e-02
## REACTOME_INSULIN_SYNTHESIS_AND_SECRETION 2.522014e-07
## REACTOME_REGULATION_OF_INSULIN_LIKE_GROWTH_FACTOR_ACTIVITY_BY_INSULIN_LIKE_GROWTH_FACTOR_BINDING_PROTEINS 1.000000e+00
## REACTOME_REGULATION_OF_INSULIN_SECRETION 3.897737e-02
## REACTOME_REGULATION_OF_INSULIN_SECRETION_BY_ACETYLCHOLINE 1.077909e-04
## REACTOME_REGULATION_OF_INSULIN_SECRETION_BY_GLUCAGON_LIKE_PEPTIDE_1 9.083131e-06
## REACTOME_REGULATION_OF_INSULIN_SECRETION_BY_FREE_FATTY_ACIDS 2.897170e-05
## REACTOME_INHIBITION_OF_INSULIN_SECRETION_BY_ADRENALINE_NORADRENALINE 1.628147e-02
## BH_pvalue
## KEGG_INSULIN_SIGNALING_PATHWAY 1.0000000000
## BIOCARTA_INSULIN_PATHWAY 0.9987657450
## REACTOME_GLUCOSE_REGULATION_OF_INSULIN_SECRETION 0.7062802680
## REACTOME_INSULIN_SYNTHESIS_AND_SECRETION 0.0002100838
## REACTOME_REGULATION_OF_INSULIN_LIKE_GROWTH_FACTOR_ACTIVITY_BY_INSULIN_LIKE_GROWTH_FACTOR_BINDING_PROTEINS 1.0000000000
## REACTOME_REGULATION_OF_INSULIN_SECRETION 0.5797883996
## REACTOME_REGULATION_OF_INSULIN_SECRETION_BY_ACETYLCHOLINE 0.0128271210
## REACTOME_REGULATION_OF_INSULIN_SECRETION_BY_GLUCAGON_LIKE_PEPTIDE_1 0.0037831242
## REACTOME_REGULATION_OF_INSULIN_SECRETION_BY_FREE_FATTY_ACIDS 0.0070612576
## REACTOME_INHIBITION_OF_INSULIN_SECRETION_BY_ADRENALINE_NORADRENALINE 0.3686245089Pathway plotting
We know that there are significantly enriched pathways in insulin secretion, so let’s plot those.
secprots <- ora.res["REACTOME_GLUCOSE_REGULATION_OF_INSULIN_SECRETION", ] |>
dplyr::select(ingroupnames) |>
unlist() |>
strsplit(split = ", ") |>
unlist()
spe.plot <- img.spes[[2]]
hup <- spatial_heatmap(spe.plot,
feature = secprots,
sample_id = "Image_1",
image_id = "with_grid",
feature_type = "PrimaryGeneName",
label_column = "IsletOrNot",
plot_title = "Glucose regulation proteins",
interactive = FALSE
)
hup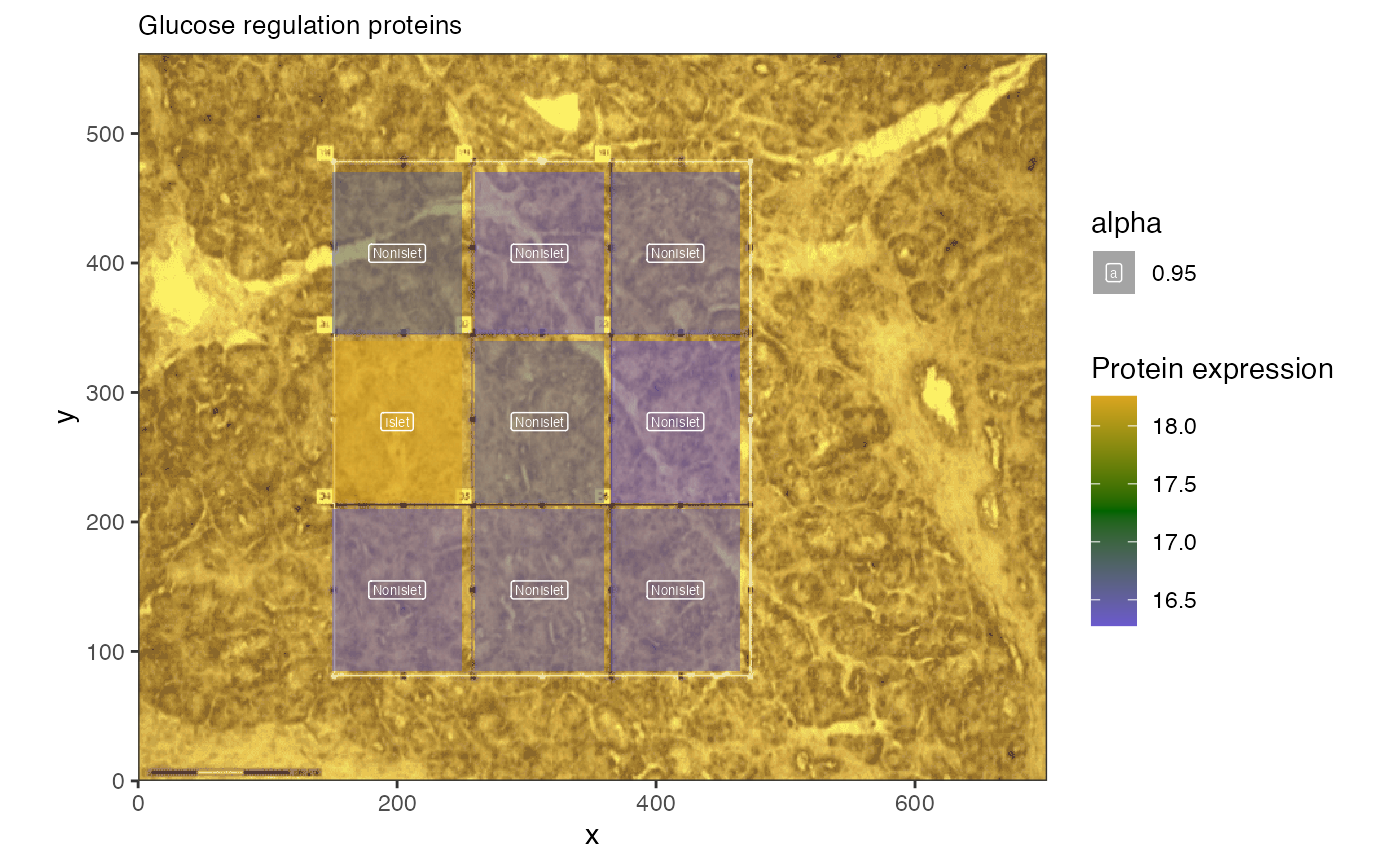
The average expression of the 20 proteins selected is shown to be higher in islet cells than adjacent cells.
Distance based measurements
We can also identify features that are correlated with distance to a feature or a gradient in the sample. This will provide input to rank-based statistical tools that can help identify pathways.
Distance based measurements
First we identify a specific feature, the Islet cell, and use that to identify proteins correlated with distance from the islet in each image. Proteins with a negative correlation are decreasing in expression as they are farther from the islet cells.
## for each image, let's compute the distance of each voxel to
## the one labeled 'Islet'
img.rank <- distance_based_analysis(img.spes[[3]], "proteomics",
sampleCategoryCol = "IsletOrNot",
sampleCategoryValue = "Islet"
)
## now we have the distances, let's plot some interesting proteins
negProts <- rowData(img.rank) |>
subset(IsletDistancespearmanPval < 0.01) |>
as.data.frame() |>
dplyr::arrange(IsletDistancespearmanCor)
print(head(negProts))## pancProts Entry EntryName
## sp|P37108|SRP14_HUMAN sp|P37108|SRP14_HUMAN P37108 SRP14_HUMAN
## sp|P52209|6PGD_HUMAN sp|P52209|6PGD_HUMAN P52209 6PGD_HUMAN
## sp|P78560|CRADD_HUMAN sp|P78560|CRADD_HUMAN P78560 CRADD_HUMAN
## sp|Q14019|COTL1_HUMAN sp|Q14019|COTL1_HUMAN Q14019 COTL1_HUMAN
## sp|Q9Y6X5|ENPP4_HUMAN sp|Q9Y6X5|ENPP4_HUMAN Q9Y6X5 ENPP4_HUMAN
## sp|P50552|VASP_HUMAN sp|P50552|VASP_HUMAN P50552 VASP_HUMAN
## ProteinNames
## sp|P37108|SRP14_HUMAN Signal recognition particle 14 kDa protein (SRP14) (18 kDa Alu RNA-binding protein)
## sp|P52209|6PGD_HUMAN 6-phosphogluconate dehydrogenase, decarboxylating (EC 1.1.1.44)
## sp|P78560|CRADD_HUMAN Death domain-containing protein CRADD (Caspase and RIP adapter with death domain) (RIP-associated protein with a death domain)
## sp|Q14019|COTL1_HUMAN Coactosin-like protein
## sp|Q9Y6X5|ENPP4_HUMAN Bis(5'-adenosyl)-triphosphatase ENPP4 (EC 3.6.1.29) (AP3A hydrolase) (AP3Aase) (Ectonucleotide pyrophosphatase/phosphodiesterase family member 4) (E-NPP 4) (NPP-4)
## sp|P50552|VASP_HUMAN Vasodilator-stimulated phosphoprotein (VASP)
## GeneNames PrimaryGeneName
## sp|P37108|SRP14_HUMAN SRP14 SRP14
## sp|P52209|6PGD_HUMAN PGD PGDH PGD
## sp|P78560|CRADD_HUMAN CRADD RAIDD CRADD
## sp|Q14019|COTL1_HUMAN COTL1 CLP COTL1
## sp|Q9Y6X5|ENPP4_HUMAN ENPP4 KIAA0879 NPP4 ENPP4
## sp|P50552|VASP_HUMAN VASP VASP
## IsletDistancespearmanCor IsletDistancespearmanPval
## sp|P37108|SRP14_HUMAN -0.9621024 3.361762e-05
## sp|P52209|6PGD_HUMAN -0.9621024 3.361762e-05
## sp|P78560|CRADD_HUMAN -0.9518763 2.686633e-04
## sp|Q14019|COTL1_HUMAN -0.9518763 2.686633e-04
## sp|Q9Y6X5|ENPP4_HUMAN -0.9518763 2.686633e-04
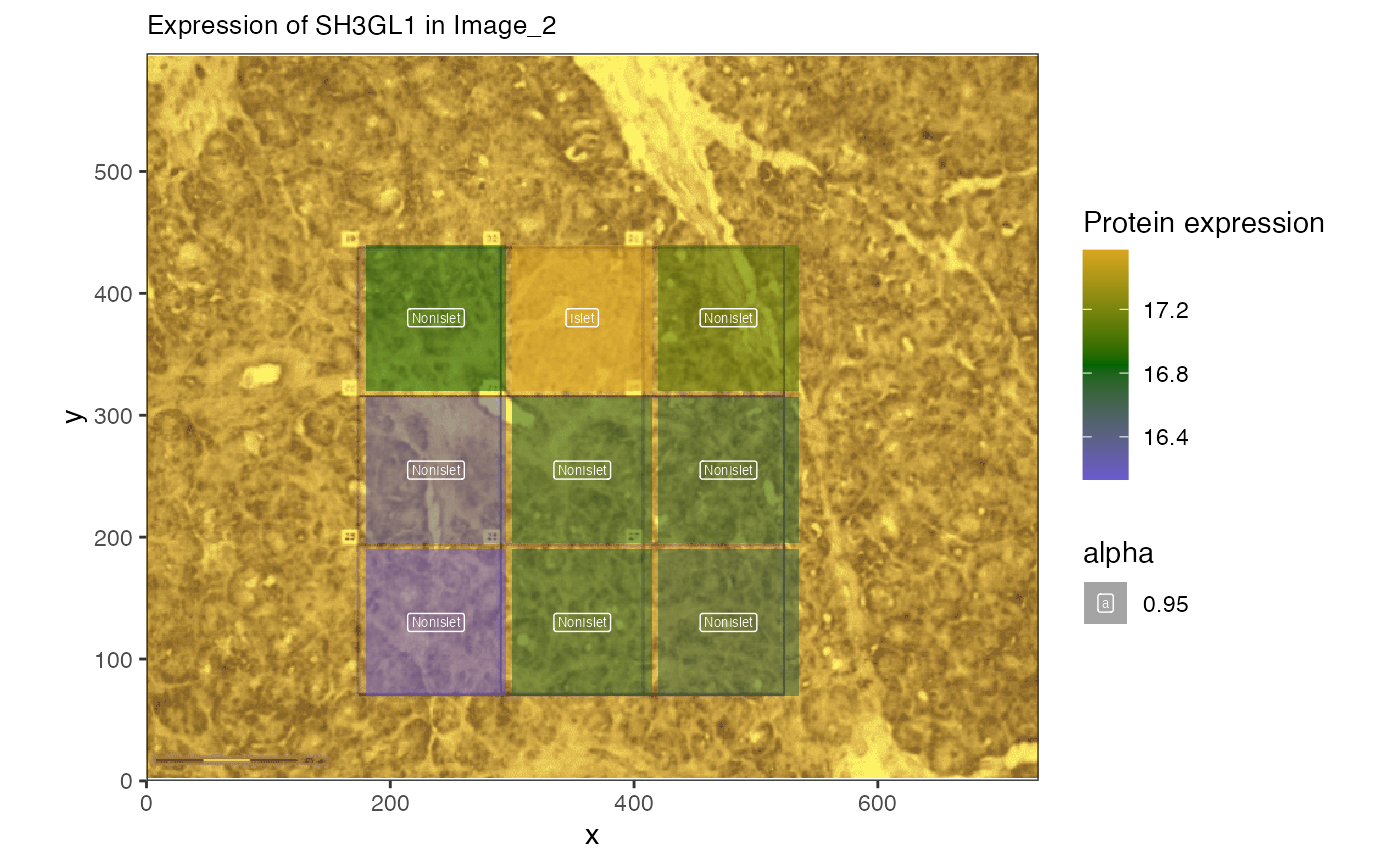
## sp|P50552|VASP_HUMAN -0.9367839 1.965329e-04It looks like SH3GL1 is correlated with distance to Islet in a few images.
Plot protein gradient
Now we can plot the expression of a protein suspected to have decreasing expression farther from the islet cells.We start with SRP14 and ENPP4
spatial_heatmap(img.spes[[3]],
feature = "SRP14",
feature_type = "PrimaryGeneName",
sample_id = names(img.spes)[3],
image_id = "with_grid",
label_column = "IsletOrNot", interactive = FALSE
)
spatial_heatmap(img.spes[[3]],
feature = "ENPP4",
feature_type = "PrimaryGeneName",
sample_id = names(img.spes)[3],
image_id = "with_grid",
label_column = "IsletOrNot", interactive = FALSE
)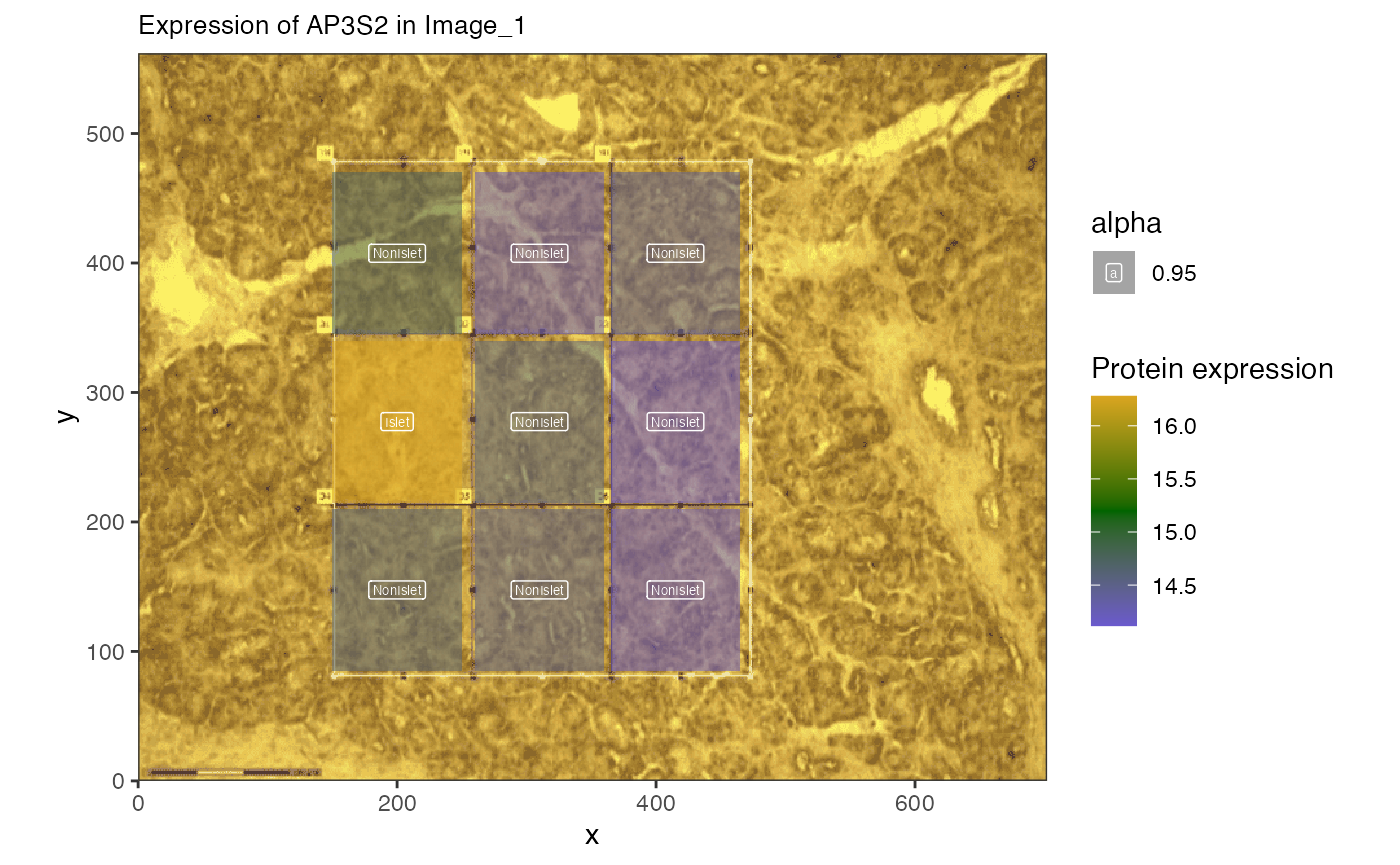
The expression of this protein is lower farther from the Islet. Can we identify trends in the proteins?
Gradient-based enrichment
Rank-based pathway enrichment is a way to evaluate trends pathways
that are over-represented in a ranked list of genes. The
leapR pathway has such functionality and we can use the
rankings as input.
library(leapR)
data("krbpaths")
spe <- img.rank
enriched.paths <- enrich_gradient(spe,
geneset = krbpaths,
method = 'ks',
feature_column = "PrimaryGeneName", # mapped to enrichment data
ranking_column = "IsletDistancespearmanCor"
)
enriched.paths[, "comp"] <- rep(names(img.spes)[[3]], nrow(enriched.paths))
enriched.paths[, "krbpaths"] <- rownames(enriched.paths)
enriched.paths |>
subset(pvalue < 0.01) |>
dplyr::arrange(BH_pvalue)## ingroup_n
## BIOCARTA_NFKB_PATHWAY 12
## KEGG_APOPTOSIS 38
## REACTOME_CYCLIN_E_ASSOCIATED_EVENTS_DURING_G1_S_TRANSITION_ 46
## KEGG_CITRATE_CYCLE_TCA_CYCLE 26
## BIOCARTA_STRESS_PATHWAY 15
## REACTOME_REGULATION_OF_APC_ACTIVATORS_BETWEEN_G1_S_AND_EARLY_ANAPHASE 51
## REACTOME_REGULATION_OF_ORNITHINE_DECARBOXYLASE 42
## KEGG_PROXIMAL_TUBULE_BICARBONATE_RECLAMATION 16
## KEGG_NEUROTROPHIN_SIGNALING_PATHWAY 59
## REACTOME_ACTIVATION_OF_BH3_ONLY_PROTEINS 8
## REACTOME_SCF_SKP2_MEDIATED_DEGRADATION_OF_P27_P21 43
## KEGG_VASCULAR_SMOOTH_MUSCLE_CONTRACTION 42
## REACTOME_CDC20_PHOSPHO_APC_MEDIATED_DEGRADATION_OF_CYCLIN_A 48
## ingroupnames
## BIOCARTA_NFKB_PATHWAY CHUK, NFKB1, NFKBIA, IRAK1, RELA, RIPK1, TRADD, TAB1, MYD88, TRAF6, IKBKG, IKBKB, MAP3K7, FADD, IL1A
## KEGG_APOPTOSIS DFFA, PIK3R2, CHUK, ENDOD1, AIFM1, CAPN1, PRKAR1A, PRKAR2A, PPP3CB, PRKACA, CAPN2, NFKB1, PRKACB, NFKBIA, PIK3R1, PRKAR2B, AKT1, AKT2, IRAK1, BID, PPP3R1, CYCS, RELA, BAX, BCL2L1, PPP3CA, TRAF2, ATM, BIRC2, RIPK1, ENDOG, TRADD, BAD, MYD88, IRAK4, IKBKG, IKBKB, CASP7, CASP8, CASP10, PRKACG, PIK3R3, PPP3CC, FADD, EXOG, IL1A, CASP6, PIK3CB
## REACTOME_CYCLIN_E_ASSOCIATED_EVENTS_DURING_G1_S_TRANSITION_ PSMD11, PSMD12, PSMD9, PSMD14, PSMA7, PSMD3, PSMD10, UBB, PSMC3, PSMB1, CDK2, PSMA1, PSMA2, PSMA3, PSMA4, PSMB8, PSMB9, PSMA5, PSMB4, PSMB6, PSMB5, PSMC2, PSMB10, PSMC4, PSMD8, PSMB3, PSMB2, PSMD7, CCNH, MNAT1, PSMD4, PSMA6, PSME3, PSMC1, PSMC5, PSMC6, SKP1, PSME1, PSMD2, CUL1, PSMD6, PSMD5, PSMF1, PSMB7, PSMD1, PSME2, PSMD13, RB1, CDK7
## KEGG_CITRATE_CYCLE_TCA_CYCLE SDHD, IDH3B, CS, IDH1, FH, PDHA1, DLD, DLAT, PDHB, PC, SDHB, SDHA, PCK1, DLST, MDH1, MDH2, IDH2, IDH3A, IDH3G, ACLY, SUCLG1, OGDH, PCK2, SUCLG2, SDHC, ACO2, SUCLA2, OGDHL
## BIOCARTA_STRESS_PATHWAY CHUK, JUN, NFKB1, NFKBIA, MAPK8, MAP2K4, MAP2K3, MAP2K6, CRADD, RELA, TRAF2, RIPK1, TRADD, MAPK14, IKBKG, IKBKB, ATF1, TANK
## REACTOME_REGULATION_OF_APC_ACTIVATORS_BETWEEN_G1_S_AND_EARLY_ANAPHASE PSMD11, PSMD12, PSMD9, PSMD14, PSMA7, PSMD3, BUB3, PSMD10, CDK1, UBB, PSMC3, PSMB1, CDK2, PSMA1, PSMA2, PSMA3, PSMA4, PSMB8, PSMB9, PSMA5, PSMB4, PSMB6, PSMB5, CDC27, PSMC2, PSMB10, PSMC4, PSMD8, PSMB3, PSMB2, PSMD7, PSMD4, PSMA6, PSME3, PSMC1, PSMC5, PSMC6, SKP1, PSME1, CDC16, PSMD2, MAD2L1, CUL1, PSMD6, PSMD5, PSMF1, PSMB7, PSMD1, CDC23, PSME2, PSMD13, UBE2E1, ANAPC1, ANAPC7, ANAPC2, ANAPC10, UBE2D1
## REACTOME_REGULATION_OF_ORNITHINE_DECARBOXYLASE PSMD11, PSMD12, PSMD9, PSMD14, PSMA7, PSMD3, PSMD10, NQO1, PSMC3, PSMB1, PSMA1, PSMA2, PSMA3, PSMA4, PSMB8, PSMB9, PSMA5, PSMB4, PSMB6, PSMB5, PSMC2, PSMB10, PSMC4, PSMD8, PSMB3, PSMB2, PSMD7, OAZ1, PSMD4, PSMA6, PSME3, PSMC1, PSMC5, PSMC6, PSME1, PSMD2, PSMD6, PSMD5, PSMF1, PSMB7, PSMD1, PSME2, PSMD13
## KEGG_PROXIMAL_TUBULE_BICARBONATE_RECLAMATION GLS, GLUD1, CA2, ATP1A1, ATP1B1, CA4, AQP1, PCK1, MDH1, GLUD2, ATP1B3, FXYD2, PCK2, SLC38A3, SLC25A10, GLS2, SLC4A4, ATP1A3
## KEGG_NEUROTROPHIN_SIGNALING_PATHWAY PIK3R2, MAP2K7, MAPK13, RIPK2, NRAS, RAF1, JUN, CALM1, NFKB1, NFKBIA, YWHAQ, MAPK3, PIK3R1, MAPK1, SHC1, AKT1, AKT2, YWHAB, MAP2K2, CSK, MAPK8, MAPK9, CRK, CRKL, MAPKAPK2, PSEN1, GSK3B, IRAK1, RPS6KA3, ARHGDIA, ARHGDIB, CDC42, RAP1B, RHOA, YWHAG, YWHAE, GRB2, RAC1, YWHAZ, MAP2K1, RELA, YWHAH, PRKCD, PTPN11, BAX, CAMK2B, CAMK2G, CAMK2D, PDK1, RPS6KA2, RPS6KA1, NFKBIB, MAPK14, BAD, SORT1, MAP3K5, IRAK4, RPS6KA6, KIDINS220, TRAF6, IKBKB, FOXO3, HRAS, NGFR, BRAF, CALML3, RAP1A, CALML5, MAGED1, PLCG2, PIK3R3, NFKBIE, PIK3CB, KRAS, PLCG1
## REACTOME_ACTIVATION_OF_BH3_ONLY_PROTEINS AKT1, YWHAB, MAPK8, BID, PPP3R1, DYNLL1, BAD, DYNLL2, PPP3CC
## REACTOME_SCF_SKP2_MEDIATED_DEGRADATION_OF_P27_P21 PSMD11, PSMD12, PSMD9, PSMD14, PSMA7, PSMD3, PSMD10, UBB, PSMC3, PSMB1, CDK2, PSMA1, PSMA2, PSMA3, PSMA4, PSMB8, PSMB9, PSMA5, PSMB4, PSMB6, PSMB5, PSMC2, PSMB10, PSMC4, PSMD8, PSMB3, PSMB2, PSMD7, PSMD4, PSMA6, PSME3, PSMC1, PSMC5, PSMC6, SKP1, PSME1, PSMD2, CUL1, PSMD6, PSMD5, PSMF1, PSMB7, PSMD1, PSME2, PSMD13
## KEGG_VASCULAR_SMOOTH_MUSCLE_CONTRACTION PPP1R12A, ROCK2, GNAS, RAF1, PLA2G1B, CALM1, ARAF, MYL6B, PRKCA, PRKACA, PRKACB, MYL9, MAPK3, MAPK1, GNA11, GUCY1A2, MYH11, MAP2K2, PPP1CC, GNAQ, MYL6, RHOA, PPP1CA, PPP1CB, ACTA2, PLCB3, MAP2K1, GNA12, PRKCD, CALD1, ROCK1, PRKG1, GNA13, ITPR2, ITPR1, MYLK, ARHGEF1, PPP1R14A, PLCB1, ARHGEF12, BRAF, CALML3, ITPR3, CALML5, PLA2G2D, PRKACG, PPP1R12B, PRKCE, PLCB4, ARHGEF11, ADCY6
## REACTOME_CDC20_PHOSPHO_APC_MEDIATED_DEGRADATION_OF_CYCLIN_A PSMD11, PSMD12, PSMD9, PSMD14, PSMA7, PSMD3, BUB3, PSMD10, CDK1, UBB, PSMC3, PSMB1, PSMA1, PSMA2, PSMA3, PSMA4, PSMB8, PSMB9, PSMA5, PSMB4, PSMB6, PSMB5, CDC27, PSMC2, PSMB10, PSMC4, PSMD8, PSMB3, PSMB2, PSMD7, PSMD4, PSMA6, PSME3, PSMC1, PSMC5, PSMC6, PSME1, CDC16, PSMD2, MAD2L1, PSMD6, PSMD5, PSMF1, PSMB7, PSMD1, CDC23, PSME2, PSMD13, UBE2E1, ANAPC1, ANAPC7, ANAPC2, ANAPC10, UBE2D1
## ingroup_mean
## BIOCARTA_NFKB_PATHWAY -0.9666485
## KEGG_APOPTOSIS -0.4769082
## REACTOME_CYCLIN_E_ASSOCIATED_EVENTS_DURING_G1_S_TRANSITION_ -0.4340674
## KEGG_CITRATE_CYCLE_TCA_CYCLE -0.5586684
## BIOCARTA_STRESS_PATHWAY -0.5554352
## REACTOME_REGULATION_OF_APC_ACTIVATORS_BETWEEN_G1_S_AND_EARLY_ANAPHASE -0.4333342
## REACTOME_REGULATION_OF_ORNITHINE_DECARBOXYLASE -0.3872065
## KEGG_PROXIMAL_TUBULE_BICARBONATE_RECLAMATION -0.5898146
## KEGG_NEUROTROPHIN_SIGNALING_PATHWAY -0.3309119
## REACTOME_ACTIVATION_OF_BH3_ONLY_PROTEINS -0.9133547
## REACTOME_SCF_SKP2_MEDIATED_DEGRADATION_OF_P27_P21 -0.4132140
## KEGG_VASCULAR_SMOOTH_MUSCLE_CONTRACTION -0.3402784
## REACTOME_CDC20_PHOSPHO_APC_MEDIATED_DEGRADATION_OF_CYCLIN_A -0.3990574
## outgroup_n
## BIOCARTA_NFKB_PATHWAY 6653
## KEGG_APOPTOSIS 6653
## REACTOME_CYCLIN_E_ASSOCIATED_EVENTS_DURING_G1_S_TRANSITION_ 6653
## KEGG_CITRATE_CYCLE_TCA_CYCLE 6653
## BIOCARTA_STRESS_PATHWAY 6653
## REACTOME_REGULATION_OF_APC_ACTIVATORS_BETWEEN_G1_S_AND_EARLY_ANAPHASE 6653
## REACTOME_REGULATION_OF_ORNITHINE_DECARBOXYLASE 6653
## KEGG_PROXIMAL_TUBULE_BICARBONATE_RECLAMATION 6653
## KEGG_NEUROTROPHIN_SIGNALING_PATHWAY 6653
## REACTOME_ACTIVATION_OF_BH3_ONLY_PROTEINS 6653
## REACTOME_SCF_SKP2_MEDIATED_DEGRADATION_OF_P27_P21 6653
## KEGG_VASCULAR_SMOOTH_MUSCLE_CONTRACTION 6653
## REACTOME_CDC20_PHOSPHO_APC_MEDIATED_DEGRADATION_OF_CYCLIN_A 6653
## outgroup_mean
## BIOCARTA_NFKB_PATHWAY 0.002190291
## KEGG_APOPTOSIS 0.003438807
## REACTOME_CYCLIN_E_ASSOCIATED_EVENTS_DURING_G1_S_TRANSITION_ 0.003794583
## KEGG_CITRATE_CYCLE_TCA_CYCLE 0.002749977
## BIOCARTA_STRESS_PATHWAY 0.001574065
## REACTOME_REGULATION_OF_APC_ACTIVATORS_BETWEEN_G1_S_AND_EARLY_ANAPHASE 0.004203927
## REACTOME_REGULATION_OF_ORNITHINE_DECARBOXYLASE 0.003088240
## KEGG_PROXIMAL_TUBULE_BICARBONATE_RECLAMATION 0.001783264
## KEGG_NEUROTROPHIN_SIGNALING_PATHWAY 0.003719528
## REACTOME_ACTIVATION_OF_BH3_ONLY_PROTEINS 0.001378649
## REACTOME_SCF_SKP2_MEDIATED_DEGRADATION_OF_P27_P21 0.003374777
## KEGG_VASCULAR_SMOOTH_MUSCLE_CONTRACTION 0.002713956
## REACTOME_CDC20_PHOSPHO_APC_MEDIATED_DEGRADATION_OF_CYCLIN_A 0.003641589
## zscore
## BIOCARTA_NFKB_PATHWAY -1.2100054
## KEGG_APOPTOSIS -0.5550717
## REACTOME_CYCLIN_E_ASSOCIATED_EVENTS_DURING_G1_S_TRANSITION_ -0.4487281
## KEGG_CITRATE_CYCLE_TCA_CYCLE -0.6356013
## BIOCARTA_STRESS_PATHWAY -0.4717361
## REACTOME_REGULATION_OF_APC_ACTIVATORS_BETWEEN_G1_S_AND_EARLY_ANAPHASE -0.4275025
## REACTOME_REGULATION_OF_ORNITHINE_DECARBOXYLASE -0.4063364
## KEGG_PROXIMAL_TUBULE_BICARBONATE_RECLAMATION -0.6804846
## KEGG_NEUROTROPHIN_SIGNALING_PATHWAY -0.2950585
## REACTOME_ACTIVATION_OF_BH3_ONLY_PROTEINS -1.1230686
## REACTOME_SCF_SKP2_MEDIATED_DEGRADATION_OF_P27_P21 -0.4191580
## KEGG_VASCULAR_SMOOTH_MUSCLE_CONTRACTION -0.3932963
## REACTOME_CDC20_PHOSPHO_APC_MEDIATED_DEGRADATION_OF_CYCLIN_A -0.3963246
## oddsratio
## BIOCARTA_NFKB_PATHWAY -0.352006404
## KEGG_APOPTOSIS -0.177630591
## REACTOME_CYCLIN_E_ASSOCIATED_EVENTS_DURING_G1_S_TRANSITION_ -0.035029623
## KEGG_CITRATE_CYCLE_TCA_CYCLE -0.109121028
## BIOCARTA_STRESS_PATHWAY -0.174363721
## REACTOME_REGULATION_OF_APC_ACTIVATORS_BETWEEN_G1_S_AND_EARLY_ANAPHASE -0.069925263
## REACTOME_REGULATION_OF_ORNITHINE_DECARBOXYLASE 0.007407511
## KEGG_PROXIMAL_TUBULE_BICARBONATE_RECLAMATION -0.165198973
## KEGG_NEUROTROPHIN_SIGNALING_PATHWAY -0.123566497
## REACTOME_ACTIVATION_OF_BH3_ONLY_PROTEINS -0.274408753
## REACTOME_SCF_SKP2_MEDIATED_DEGRADATION_OF_P27_P21 -0.011521398
## KEGG_VASCULAR_SMOOTH_MUSCLE_CONTRACTION -0.097412306
## REACTOME_CDC20_PHOSPHO_APC_MEDIATED_DEGRADATION_OF_CYCLIN_A -0.062994096
## pvalue
## BIOCARTA_NFKB_PATHWAY 0.001038605
## KEGG_APOPTOSIS 0.003005928
## REACTOME_CYCLIN_E_ASSOCIATED_EVENTS_DURING_G1_S_TRANSITION_ 0.003978543
## KEGG_CITRATE_CYCLE_TCA_CYCLE 0.004268044
## BIOCARTA_STRESS_PATHWAY 0.004632733
## REACTOME_REGULATION_OF_APC_ACTIVATORS_BETWEEN_G1_S_AND_EARLY_ANAPHASE 0.004641338
## REACTOME_REGULATION_OF_ORNITHINE_DECARBOXYLASE 0.005209758
## KEGG_PROXIMAL_TUBULE_BICARBONATE_RECLAMATION 0.006106393
## KEGG_NEUROTROPHIN_SIGNALING_PATHWAY 0.006361512
## REACTOME_ACTIVATION_OF_BH3_ONLY_PROTEINS 0.008037105
## REACTOME_SCF_SKP2_MEDIATED_DEGRADATION_OF_P27_P21 0.008192979
## KEGG_VASCULAR_SMOOTH_MUSCLE_CONTRACTION 0.008713546
## REACTOME_CDC20_PHOSPHO_APC_MEDIATED_DEGRADATION_OF_CYCLIN_A 0.009351959
## BH_pvalue
## BIOCARTA_NFKB_PATHWAY 0.3809652
## KEGG_APOPTOSIS 0.3809652
## REACTOME_CYCLIN_E_ASSOCIATED_EVENTS_DURING_G1_S_TRANSITION_ 0.3809652
## KEGG_CITRATE_CYCLE_TCA_CYCLE 0.3809652
## BIOCARTA_STRESS_PATHWAY 0.3809652
## REACTOME_REGULATION_OF_APC_ACTIVATORS_BETWEEN_G1_S_AND_EARLY_ANAPHASE 0.3809652
## REACTOME_REGULATION_OF_ORNITHINE_DECARBOXYLASE 0.3809652
## KEGG_PROXIMAL_TUBULE_BICARBONATE_RECLAMATION 0.3809652
## KEGG_NEUROTROPHIN_SIGNALING_PATHWAY 0.3809652
## REACTOME_ACTIVATION_OF_BH3_ONLY_PROTEINS 0.3809652
## REACTOME_SCF_SKP2_MEDIATED_DEGRADATION_OF_P27_P21 0.3809652
## KEGG_VASCULAR_SMOOTH_MUSCLE_CONTRACTION 0.3809652
## REACTOME_CDC20_PHOSPHO_APC_MEDIATED_DEGRADATION_OF_CYCLIN_A 0.3809652
## SignedBH_pvalue
## BIOCARTA_NFKB_PATHWAY -0.3809652
## KEGG_APOPTOSIS -0.3809652
## REACTOME_CYCLIN_E_ASSOCIATED_EVENTS_DURING_G1_S_TRANSITION_ -0.3809652
## KEGG_CITRATE_CYCLE_TCA_CYCLE -0.3809652
## BIOCARTA_STRESS_PATHWAY -0.3809652
## REACTOME_REGULATION_OF_APC_ACTIVATORS_BETWEEN_G1_S_AND_EARLY_ANAPHASE -0.3809652
## REACTOME_REGULATION_OF_ORNITHINE_DECARBOXYLASE -0.3809652
## KEGG_PROXIMAL_TUBULE_BICARBONATE_RECLAMATION -0.3809652
## KEGG_NEUROTROPHIN_SIGNALING_PATHWAY -0.3809652
## REACTOME_ACTIVATION_OF_BH3_ONLY_PROTEINS -0.3809652
## REACTOME_SCF_SKP2_MEDIATED_DEGRADATION_OF_P27_P21 -0.3809652
## KEGG_VASCULAR_SMOOTH_MUSCLE_CONTRACTION -0.3809652
## REACTOME_CDC20_PHOSPHO_APC_MEDIATED_DEGRADATION_OF_CYCLIN_A -0.3809652
## background_n
## BIOCARTA_NFKB_PATHWAY NA
## KEGG_APOPTOSIS NA
## REACTOME_CYCLIN_E_ASSOCIATED_EVENTS_DURING_G1_S_TRANSITION_ NA
## KEGG_CITRATE_CYCLE_TCA_CYCLE NA
## BIOCARTA_STRESS_PATHWAY NA
## REACTOME_REGULATION_OF_APC_ACTIVATORS_BETWEEN_G1_S_AND_EARLY_ANAPHASE NA
## REACTOME_REGULATION_OF_ORNITHINE_DECARBOXYLASE NA
## KEGG_PROXIMAL_TUBULE_BICARBONATE_RECLAMATION NA
## KEGG_NEUROTROPHIN_SIGNALING_PATHWAY NA
## REACTOME_ACTIVATION_OF_BH3_ONLY_PROTEINS NA
## REACTOME_SCF_SKP2_MEDIATED_DEGRADATION_OF_P27_P21 NA
## KEGG_VASCULAR_SMOOTH_MUSCLE_CONTRACTION NA
## REACTOME_CDC20_PHOSPHO_APC_MEDIATED_DEGRADATION_OF_CYCLIN_A NA
## background_mean
## BIOCARTA_NFKB_PATHWAY NA
## KEGG_APOPTOSIS NA
## REACTOME_CYCLIN_E_ASSOCIATED_EVENTS_DURING_G1_S_TRANSITION_ NA
## KEGG_CITRATE_CYCLE_TCA_CYCLE NA
## BIOCARTA_STRESS_PATHWAY NA
## REACTOME_REGULATION_OF_APC_ACTIVATORS_BETWEEN_G1_S_AND_EARLY_ANAPHASE NA
## REACTOME_REGULATION_OF_ORNITHINE_DECARBOXYLASE NA
## KEGG_PROXIMAL_TUBULE_BICARBONATE_RECLAMATION NA
## KEGG_NEUROTROPHIN_SIGNALING_PATHWAY NA
## REACTOME_ACTIVATION_OF_BH3_ONLY_PROTEINS NA
## REACTOME_SCF_SKP2_MEDIATED_DEGRADATION_OF_P27_P21 NA
## KEGG_VASCULAR_SMOOTH_MUSCLE_CONTRACTION NA
## REACTOME_CDC20_PHOSPHO_APC_MEDIATED_DEGRADATION_OF_CYCLIN_A NA
## comp
## BIOCARTA_NFKB_PATHWAY Image_2
## KEGG_APOPTOSIS Image_2
## REACTOME_CYCLIN_E_ASSOCIATED_EVENTS_DURING_G1_S_TRANSITION_ Image_2
## KEGG_CITRATE_CYCLE_TCA_CYCLE Image_2
## BIOCARTA_STRESS_PATHWAY Image_2
## REACTOME_REGULATION_OF_APC_ACTIVATORS_BETWEEN_G1_S_AND_EARLY_ANAPHASE Image_2
## REACTOME_REGULATION_OF_ORNITHINE_DECARBOXYLASE Image_2
## KEGG_PROXIMAL_TUBULE_BICARBONATE_RECLAMATION Image_2
## KEGG_NEUROTROPHIN_SIGNALING_PATHWAY Image_2
## REACTOME_ACTIVATION_OF_BH3_ONLY_PROTEINS Image_2
## REACTOME_SCF_SKP2_MEDIATED_DEGRADATION_OF_P27_P21 Image_2
## KEGG_VASCULAR_SMOOTH_MUSCLE_CONTRACTION Image_2
## REACTOME_CDC20_PHOSPHO_APC_MEDIATED_DEGRADATION_OF_CYCLIN_A Image_2
## krbpaths
## BIOCARTA_NFKB_PATHWAY BIOCARTA_NFKB_PATHWAY
## KEGG_APOPTOSIS KEGG_APOPTOSIS
## REACTOME_CYCLIN_E_ASSOCIATED_EVENTS_DURING_G1_S_TRANSITION_ REACTOME_CYCLIN_E_ASSOCIATED_EVENTS_DURING_G1_S_TRANSITION_
## KEGG_CITRATE_CYCLE_TCA_CYCLE KEGG_CITRATE_CYCLE_TCA_CYCLE
## BIOCARTA_STRESS_PATHWAY BIOCARTA_STRESS_PATHWAY
## REACTOME_REGULATION_OF_APC_ACTIVATORS_BETWEEN_G1_S_AND_EARLY_ANAPHASE REACTOME_REGULATION_OF_APC_ACTIVATORS_BETWEEN_G1_S_AND_EARLY_ANAPHASE
## REACTOME_REGULATION_OF_ORNITHINE_DECARBOXYLASE REACTOME_REGULATION_OF_ORNITHINE_DECARBOXYLASE
## KEGG_PROXIMAL_TUBULE_BICARBONATE_RECLAMATION KEGG_PROXIMAL_TUBULE_BICARBONATE_RECLAMATION
## KEGG_NEUROTROPHIN_SIGNALING_PATHWAY KEGG_NEUROTROPHIN_SIGNALING_PATHWAY
## REACTOME_ACTIVATION_OF_BH3_ONLY_PROTEINS REACTOME_ACTIVATION_OF_BH3_ONLY_PROTEINS
## REACTOME_SCF_SKP2_MEDIATED_DEGRADATION_OF_P27_P21 REACTOME_SCF_SKP2_MEDIATED_DEGRADATION_OF_P27_P21
## KEGG_VASCULAR_SMOOTH_MUSCLE_CONTRACTION KEGG_VASCULAR_SMOOTH_MUSCLE_CONTRACTION
## REACTOME_CDC20_PHOSPHO_APC_MEDIATED_DEGRADATION_OF_CYCLIN_A REACTOME_CDC20_PHOSPHO_APC_MEDIATED_DEGRADATION_OF_CYCLIN_AWe can see that numerous pathways are coming up as enriched across images, including ribosomal and translation related pathways. Now we can select proteins from a particular pathway and visualize those as well.
Plotting pathways from gradient
rprots <- subset(enriched.paths, krbpaths == "REACTOME_ACTIVATION_OF_BH3_ONLY_PROTEINS") |>
dplyr::select(comp, ingroupnames)
rprots <- unlist(strsplit(rprots[1, 2], split = ", "))
spatial_heatmap(img.rank,
feature = rprots,
feature_type = "PrimaryGeneName",
sample_id = names(img.spes)[3],
image_id = "with_grid",
label_column = "IsletOrNot", interactive = FALSE
)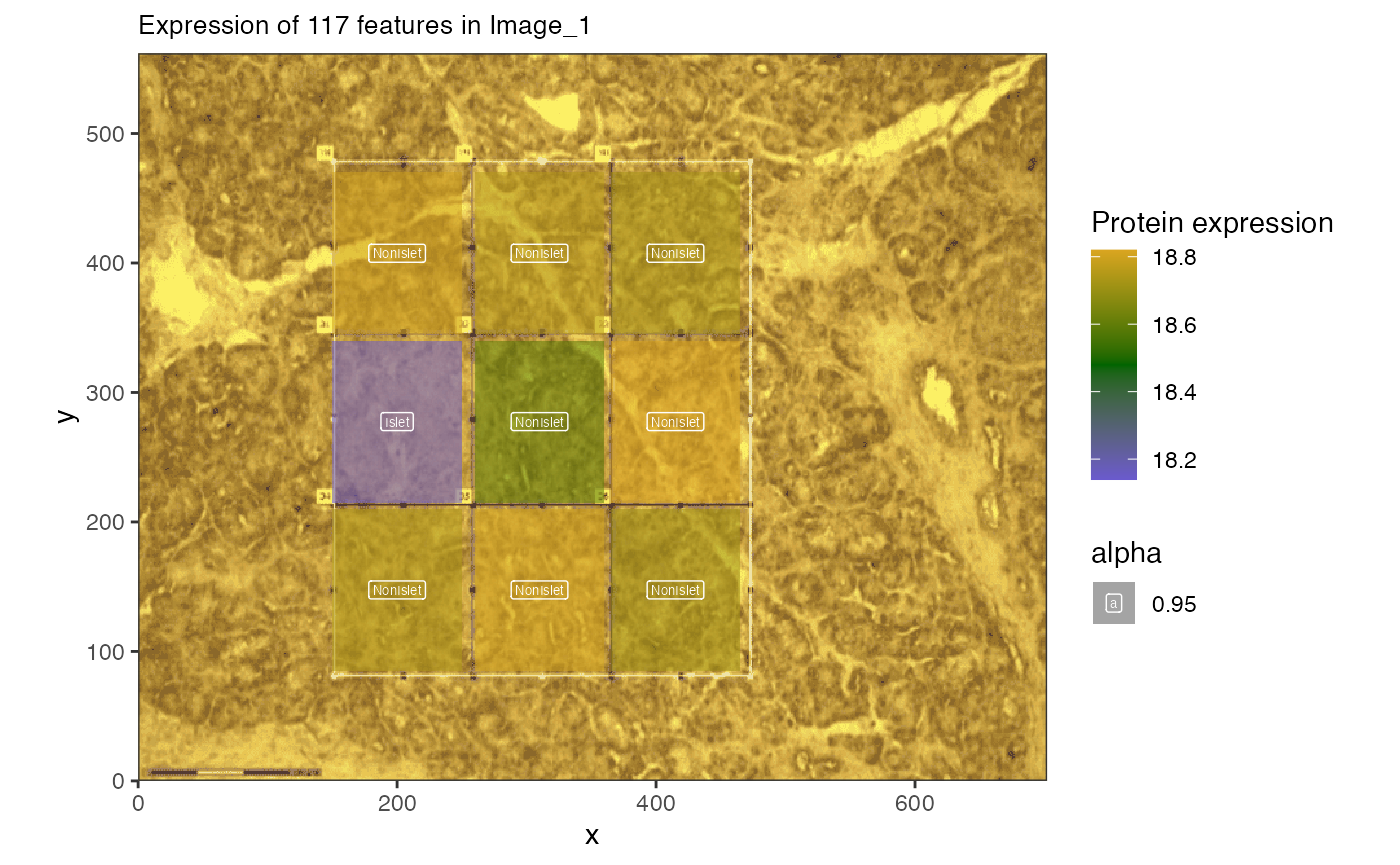
This shows the ribosomal protein expression across the image.
Network plotting
We can also look at the correlation of the ribosomal proteins in a graph. The correlation code takes a while but then we can reduce the graph to the proteins we are most interested in, or those that are most correlated.
##
## Attaching package: 'tidygraph'## The following objects are masked from 'package:IRanges':
##
## active, slice## The following objects are masked from 'package:S4Vectors':
##
## active, rename## The following object is masked from 'package:stats':
##
## filter## Loading required package: ggplot2
##correlation analysis can be slow, so let's only evaluate the top 1000 most variable proteins
varprots = apply(assay(img.spes[[3]]),1,var,na.rm = TRUE) |>
sort(decreasing = TRUE) |>
names()
full_graph <- spatial_network(img.spes[[3]],target_features = rprots,
'proteomics','PrimaryGeneName')## Joining with `by = join_by(rowval)`
##how subset for only those 81 proteins
rgraph <- full_graph |>
tidygraph::activate(nodes) |>
dplyr::filter(name %in% rprots) |>#[sample(20)]) |>
tidygraph::activate(edges) |>
dplyr::filter(abs(corval) > 0.25)
##then we can plot
ggraph::ggraph(rgraph) +
geom_edge_link(aes(colour = corval)) +
geom_node_point() +
geom_node_label(aes(label = name))## Using "stress" as default layout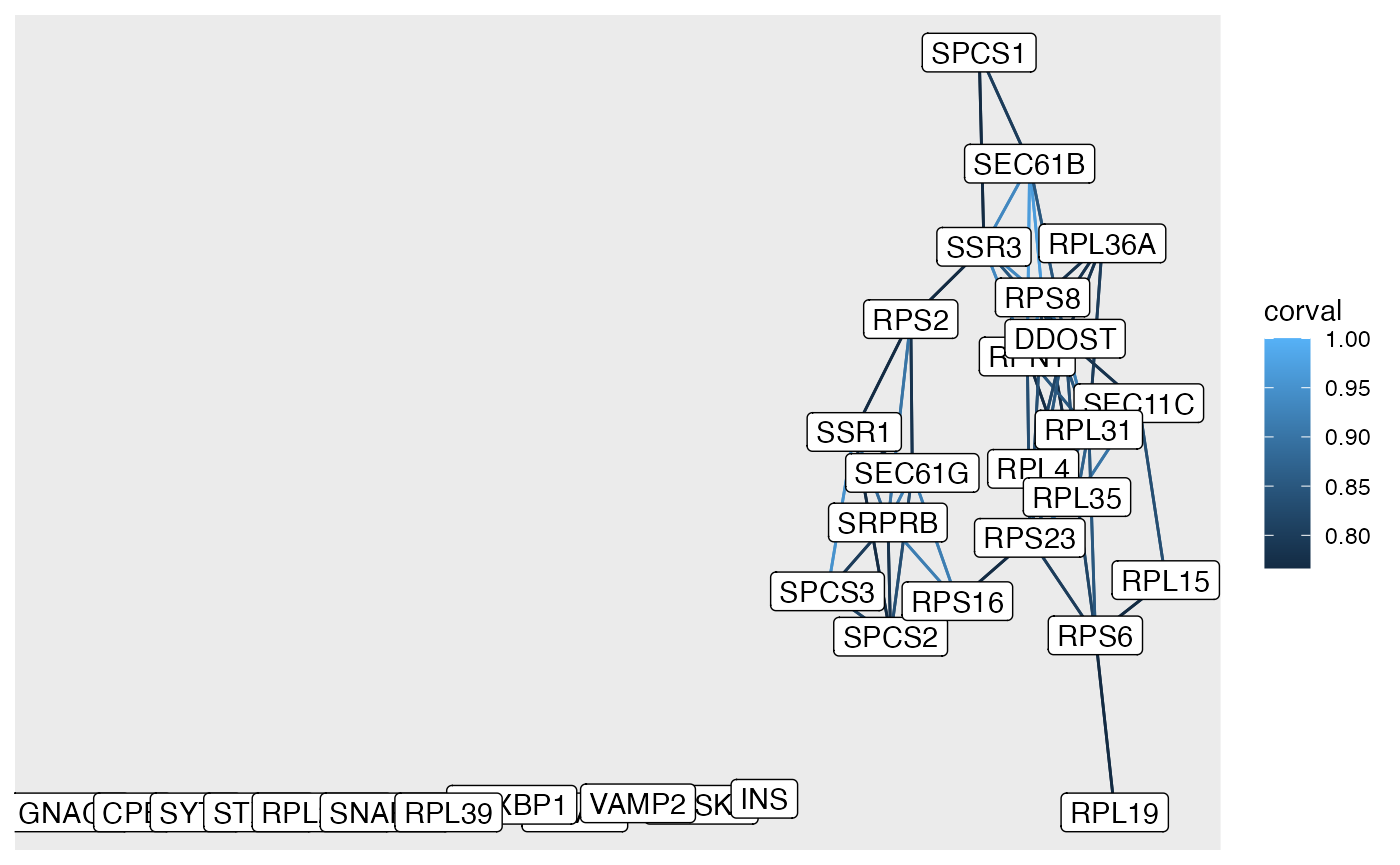 Here are the highly correlated edges between the proteins selected in
the cytokine pathway.
Here are the highly correlated edges between the proteins selected in
the cytokine pathway.
Summary
This vignette shows various functions to apply in managing spatial proteomics data in spammR.
Session info
## R version 4.6.1 (2026-06-24)
## Platform: x86_64-pc-linux-gnu
## Running under: Ubuntu 24.04.4 LTS
##
## Matrix products: default
## BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
## LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.26.so; LAPACK version 3.12.0
##
## locale:
## [1] LC_CTYPE=C.UTF-8 LC_NUMERIC=C LC_TIME=C.UTF-8
## [4] LC_COLLATE=C.UTF-8 LC_MONETARY=C.UTF-8 LC_MESSAGES=C.UTF-8
## [7] LC_PAPER=C.UTF-8 LC_NAME=C LC_ADDRESS=C
## [10] LC_TELEPHONE=C LC_MEASUREMENT=C.UTF-8 LC_IDENTIFICATION=C
##
## time zone: UTC
## tzcode source: system (glibc)
##
## attached base packages:
## [1] stats4 stats graphics grDevices utils datasets methods
## [8] base
##
## other attached packages:
## [1] ggraph_2.2.2 ggplot2_4.0.3
## [3] tidygraph_1.3.1 leapR_1.0.0
## [5] BiocFileCache_3.2.0 dbplyr_2.6.0
## [7] spammR_0.99.23 limma_3.68.4
## [9] SpatialExperiment_1.22.0 SingleCellExperiment_1.34.0
## [11] SummarizedExperiment_1.42.0 Biobase_2.72.0
## [13] GenomicRanges_1.64.0 Seqinfo_1.2.0
## [15] IRanges_2.46.0 S4Vectors_0.50.1
## [17] BiocGenerics_0.58.1 generics_0.1.4
## [19] MatrixGenerics_1.24.0 matrixStats_1.5.0
## [21] BiocStyle_2.40.0
##
## loaded via a namespace (and not attached):
## [1] RColorBrewer_1.1-3 jsonlite_2.0.0 wk_0.9.5
## [4] magrittr_2.0.5 magick_2.9.1 farver_2.1.2
## [7] rmarkdown_2.31 fs_2.1.0 ragg_1.5.2
## [10] vctrs_0.7.3 spdep_1.4-2 memoise_2.0.1
## [13] rstatix_1.0.0 htmltools_0.5.9 S4Arrays_1.12.0
## [16] curl_7.1.0 broom_1.0.13 s2_1.1.11
## [19] SparseArray_1.12.2 Formula_1.2-5 sass_0.4.10
## [22] spData_2.3.5 KernSmooth_2.23-26 bslib_0.11.0
## [25] htmlwidgets_1.6.4 desc_1.4.3 httr2_1.2.3
## [28] impute_1.86.0 plotly_4.12.0 cachem_1.1.0
## [31] igraph_2.3.3 lifecycle_1.0.5 pkgconfig_2.0.3
## [34] Matrix_1.7-5 R6_2.6.1 fastmap_1.2.0
## [37] digest_0.6.39 ggnewscale_0.5.2 textshaping_1.0.5
## [40] RSQLite_3.53.3 ggpubr_1.0.0 filelock_1.0.3
## [43] labeling_0.4.3 httr_1.4.8 polyclip_1.10-7
## [46] abind_1.4-8 compiler_4.6.1 proxy_0.4-29
## [49] bit64_4.8.2 withr_3.0.3 S7_0.2.2
## [52] backports_1.5.1 carData_3.0-6 viridis_0.6.5
## [55] DBI_1.3.0 ggforce_0.5.0 ggsignif_0.6.4
## [58] MASS_7.3-65 rappdirs_0.3.4 DelayedArray_0.38.2
## [61] rjson_0.2.23 classInt_0.4-11 tools_4.6.1
## [64] units_1.0-1 otel_0.2.0 glue_1.8.1
## [67] grid_4.6.1 sf_1.1-1 gtable_0.3.6
## [70] tzdb_0.5.0 class_7.3-23 tidyr_1.3.2
## [73] data.table_1.18.4 hms_1.1.4 sp_2.2-1
## [76] car_3.1-5 XVector_0.52.0 ggrepel_0.9.8
## [79] pillar_1.11.1 dplyr_1.2.1 tweenr_2.0.3
## [82] lattice_0.22-9 bit_4.6.0 deldir_2.0-4
## [85] tidyselect_1.2.1 knitr_1.51 gridExtra_2.3.1
## [88] bookdown_0.47 xfun_0.59 graphlayouts_1.2.4
## [91] statmod_1.5.2 lazyeval_0.2.3 yaml_2.3.12
## [94] boot_1.3-32 evaluate_1.0.5 tibble_3.3.1
## [97] BiocManager_1.30.27 cli_3.6.6 reticulate_1.46.0
## [100] systemfonts_1.3.2 jquerylib_0.1.4 Rcpp_1.1.2
## [103] png_0.1-9 pkgdown_2.2.1 readr_2.2.0
## [106] blob_1.3.0 viridisLite_0.4.3 scales_1.4.0
## [109] e1071_1.7-17 purrr_1.2.2 rlang_1.3.0